
The mcPolymer command-line interface
The mcPolymer program is used in a terminal environment. In the Windows operating system this is the Command Prompt (cmd.exe or cmd) and at Linux any command shell (e.g. tcsh, bash). The following examples assume that the mcPolymer program (Windows: mcPolymer.exe, Linux: mcPolymer) and the model file "Styrene.mcPolymer" are in the current directory.
Calling the program with the --help option you can see all the features of the command line interface and a basic example of their application.
c:\tmp\mcPolymer\mcPolymer --help
Example:
mcPolymer -m Styrene.mcPolymer --time=3600 --dt=60 --temperature=70.0 --molecules=10000000000
Usage:
mcPolymer -m modelFile [OPTION...]
-m, --model modelFILE Model
--help Print help
--license Print license
--time double reaction time / s
--dt double time interval for output concentrations / s
--dtCLD double time interval for output cld | mmd / s
--temperature double temperature / centigrade
--pressure double pressure / bar
--molecules unsigned int num moleculesThe option --license provides mcPolymer license status information. The mcPolymer simulator is published as open source software under the GNU general public license. On the web page "Download and Buid mcPolymer" the source code of the program is available for download. The following external software components are integrated in mcPolymer and are part of the distribution:
- File cxxopts.hpp contains one C++ option parser library, supporting the standard GNU style syntax for options. This is a header only library and was developed by Jarryd Beck. Source: GitHub. github.com/jarro2783/cxxopts
- File MersenneTwister.h implements the Mersenne Twister random number generator, developed by Makoto Matsumoto, Takuji Nishimura, Shawn Cokus and Richard J. Wagner. Source: GitHub. github.com/shhyang/simbats/blob/master/MersenneTwister.h
Running a simulation requires a kinetic model. This is provided in a file which name is entered in the command line with the option -m filename or --model filename. In the example shown with the --help option, the model file Styrene.mcPolymer is used for the simulation (-m Styrene.mcPolymer).
The --time option determines the reaction time in seconds running the kinetic model. This time can optionally also be specified in the model file (e.g. time=3600). The entry on the command line overrules the value in the model file. The reaction time is required for the simulation has to be specified on at least one of those two places (command line: --time=value; model file: time=value). Otherwise mcPolymer terminates with an error message: "reaction time undefined". Generally for the pattern --option=value no spaces are allowed in front of or behind the "=", otherwise the interpretation on the command line will fail.
Equally essential is the input of the reaction temperature in degrees Celsius, which can also be done in the model file (temperature=value) or on the command line (--temperature=value). The same rules as for the reaction time will also be applied here. The entry of --pressure=value is optional; 1 bar is the default pressure.
For the kinetic Monte Carlo (kMC) simulation, the definition of the sample size is required. This is done by entering the statement --molecules=value on the command line. In our example --molecules=10000000000, the 1e10 molecules are distributed to the educts according to the concentrations specified in the model file ([styrene]0 = 8.725 mol·L-1 ⇒ 9977129788 molecules, [AIBN]0 = 0.02 mol·L-1 ⇒ 22870212molecules). This corresponds to a control volume of 1.9e-15 L. It is clear that the sample size in the kMC has a significant impact on the precision of the simulation but also on computation time and memory requirements. In this example (-m Styrene.mcPolymer --time=3600 --temperature=70.0 --molecules=10000000000 ) the simulation takes about 3 minutes on an Intel Core i7-6700, 3.4 GHz CPU and requires a maximum of 530 MB of RAM.
The command line options --dt=value and --dtCLD =value control the logging of results. Both parameters have default values (dt=1/60·reaction time; dtCLD=reaction time), which are used if these options are not set. After starting the program with the options mentioned in the example, the current parameters are displayed. In addition, an interval of dt (--dt=60) seconds is used to output the current reaction time in the running kMC simulation:
c:\tmp\mcPolymer -m Styrene.mcPolymer --dt=60 --temperature=70 --molecules=10000000000 model file: Styrene.mcPolymer model dimension: 10000000000 ( 1.000000e+10 ) molecules reaction time 3600, defined in model file concentration output dt 60 s cld | mmd output dtCLD 3600 s, set to reaction time by default reaction temperature 70 degree Celsius pressure 1 bar, set by default time: 60 time: 120 time: 180 ..
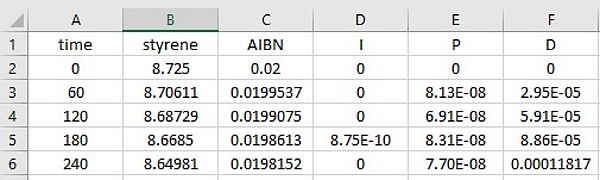
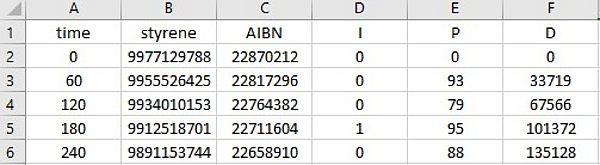
The concentration time and molecule number-time profiles are written in two files, which are updated at intervals of dt. The file names are derived from the name of the model file:
- modelfilename.Concentrations.dat (e.g. Styrene.mcPolymer.Concentrations.dat)
- modelfilename.numMolecules.dat (e.g. Styrene.mcPolymer.numMolecules.dat).
Both files are ASCII files in which the columns are separated by a tab character. This makes a data analysis or visualization in other programs easily possible. For each reaction time, the concentration (mol·L-1) or the number of molecules of each reactant contained in the kinetic model is available:
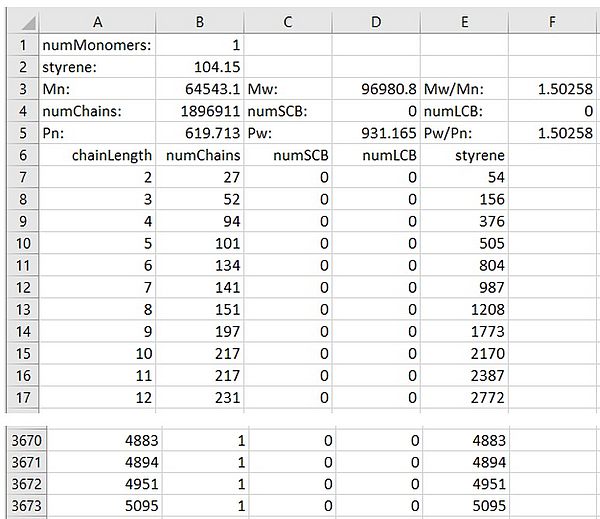
Additionally, chain length distributions and molar mass distributions of all macromolecular species defined in the kinetic model are available as output files written at intervals specified by --dtCLD. The file name contains the name of the species and the reaction time. At the end of the simulation, two ASCII files are written e.g. for the dead polymer D defined in the model Styrene.mcPolymer: chain length distribution ⇒ D.3600.000000.CLD.dat and molar mass distribution ⇒ D.3600.000000.CLD.dat.WlogM.dat. The molar mass distribution is stored in equidistant X-axis steps (log (M)) and normalized to area 1. The file with the chain length distribution has the following structure:
In the file header, the mean values and the number of polymer molecules are summarized. The chain length distribution begins in this example at line 6 and is located in columns A (chain length) and B (number of chains). In addition, each chain length is assigned the number of short chain branches (column C, numSCB) and long chain branches (column D, numLCB). In this simple example Styrol.mcPolymer, no branching reactions occur. In column E, the number of monomer units in the chains of the corresponding chain length is given. In the styrene homopolymerization, only column E is occupied. In a copolymerization, the number of these monomers is located in additional columns, starting with F.